
AI-Designed Phages
A new paper shows that a generative AI model can design viable bacteriophages.

A few months ago, Arc Institute released a new language model, called Evo 2, that can design entire genomes. In that original paper, though, the model’s designs — for a yeast chromosome and some small bacterial genomes — were entirely confined to a computer. The AI-generated genomes were not assembled or tested in the laboratory.
Although AI models are exceptionally good at designing proteins (including, recently, highly dynamic enzymes), there was little evidence that AI models could design viable genomes. Proteins are self-contained entities, made from a single strand of amino acids. But even the simplest genomes are composed of multiple genes and regulatory elements that must collaborate to build a functioning, living organism. A single mutation in a genome is often enough to render it entirely defunct.
But today, Arc Institute and Stanford University researchers have validated their designs in the real world, reporting the first viable genomes created using generative AI. They used fine-tuned versions of both Evo 1 and Evo 2 to create 16 bacteriophages modeled on ΦX174, a virus that infects E. coli bacteria.1 Some of these AI-generated phages work just as well or better at infecting E. coli cells compared to wild ΦX174. All of the fine-tuned models used in this work are also freely available on HuggingFace. The paper offers “a blueprint for the design of diverse synthetic bacteriophages,” the authors write, “and, more broadly, lays a foundation for the generative design of useful living systems at the genome scale.”
Choosing the Phage
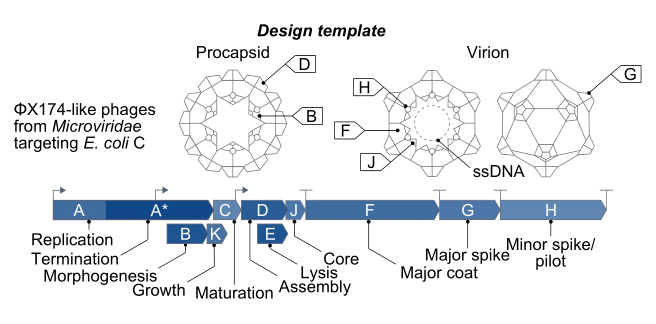
Of the 13,000 known bacteriophage types, ΦX174 is the most widely studied. First discovered in the Paris sewers in 1935, its genome includes only 5,000 bases of single-stranded DNA, with eleven genes and at least seven regulatory elements, or short stretches of DNA that regulate which genes switch on at which times. So many genes fit in such a small sequence because they physically overlap one another, with some genes tucked in the middle of other genes.
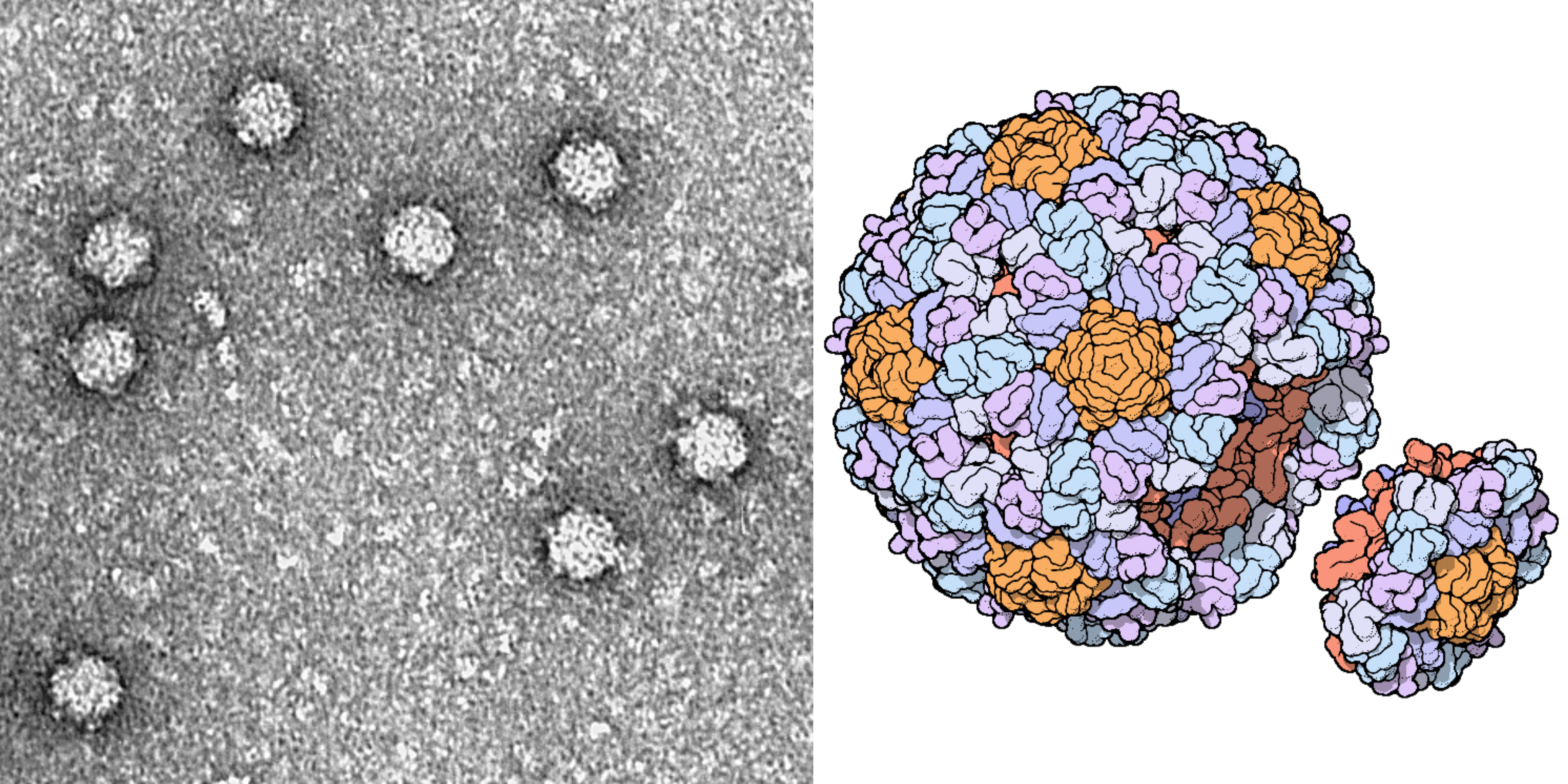
ΦX174 is often used as a model organism in molecular biology because it is easy to work with. It infects a nonpathogenic strain of E. coli, which itself divides quickly and can be readily grown in the laboratory using nutrient-laden broth and a warm incubator. These phages are also structurally simple, even by bacteriophage standards — they are made from little more than a capsid, packed with the small genome, and some proteins.
It is these proteins, embedded on the outside of each ΦX174 capsid, that latch onto E. coli cells. Once bound, the phage injects its DNA into the unwitting host, and the infected bacterium begins using its own protein machinery to replicate the phage DNA. Once the cell has made hundreds of copies, the phages assemble and burst through the host’s cell membrane, killing it. Freed from the wreckage of the lysed bacterium, the new phages disperse through the surrounding broth, colliding with other E. coli cells they can infect.
ΦX174 is also of immense historical importance. It was the first complete DNA genome ever to be sequenced (by Frederick Sanger in 1977) and chemically synthesized and assembled. In 2003, J. Craig Venter and colleagues devised a technique to “overlap” short DNA sequences to build larger ones. In just 14 days, they assembled the full genome of ΦX174 and showed that they could “boot it up” by pushing the DNA into E. coli and coaxing those cells to make infectious virions.
Finally, ΦX174 was the proving ground for one of the most famous synthetic biology papers of all time. In 2012, Paul Jaschke, Drew Endy, and others at Stanford University “decompressed” and extended the ΦX174 genome, separated out all its overlapping genes, synthesized the DNA, and showed that the genome still produced infectious phages. This was one of the earliest case studies of true “genome design,” in which a natural template was highly modified yet still retained its function.2
So even though this new preprint isn’t the first time that researchers have designed and built a synthetic genome, it is the first time an AI model has allowed them to go “off script” and create viable genomes that humans, rationally, could not. Some of the viable phage genomes created in this work are so distinct from any known bacteriophage genome that they would technically be classified as their own species.
{kind=link}
Designing the Genomes
Evo 2 is a genome language model that can “read” and “interpret” genetic sequences from all domains of life. Evo 2 was trained on 9.3 trillion nucleotides of DNA from 128,000 different organisms. Instead of words, its inputs and outputs are strings of DNA. To design new phage genomes, the researchers prompted both Evo 1 (a smaller, earlier model trained only on bacterial genomes) and Evo 2 to generate candidate sequences.
Specifically, they prompted each model with a “fixed” portion of the ΦX174 genome — a consensus sequence that is conserved across all natural isolates — and then instructed the model to “fill in” the missing stretches. The Evo models extend this prompt or “seed” sequence into complete, synthetic genomes.3
Although Evo 2, for example, was trained on millions of bacteriophage genomes, it cannot necessarily design viable phages on its own. In our prior coverage of Evo 2, we explained how the model was used to design a small bacterial genome, but that this genome “was missing some critical elements … and so would likely not function if synthesized and inserted into a real bacterium.”
Still, a phage genome is far simpler than a bacterial one. And so, in this work, the researchers first tested whether their Evo models could spit out anything even resembling one.
After prompting both models to generate candidate sequences, they ran these sequences through geNomad, a classification tool that can identify viral DNA. Just 19–33 percent of Evo 1 sequences were flagged as viral, while 34–38 percent of Evo 2 sequences were flagged in the same way. Ultimately, the base models can occasionally generate sequences that resemble bacteriophages, but not reliably — and still with no evidence that any of them would function.
To improve the odds that the outputted phages could infect living E. coli, the researchers fine-tuned Evo on a cleaned dataset of nearly 15,000 Microviridae genomes — the viral family that ΦX174 belongs to — and used it to nudge the model toward generating more realistic candidates. They also added some simple computational filters to remove any sequences that would likely be nonviable, such as “only keep sequences between 4,000 and 6,000 bases long, those that encode all the necessary genes,” and so on. After generating and filtering thousands of genomes, they were left with 302 distinct phage genome candidates.
Collectively, these remaining candidates had hundreds of unique mutations; some had an additional gene inserted, for example, or carried scrambled genes. (Paradoxically, one genome that was more than 99 percent identical to ΦX174 turned out to be non-viable, producing no living phages at all.)4
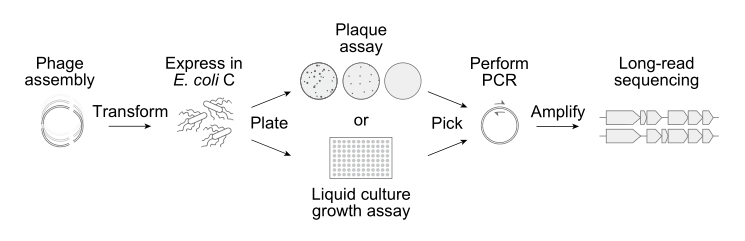
Next, the Arc team built the 302 AI-generated phage sequences in the laboratory. Only 285 of them could be chemically-synthesized and assembled,5 however, and then inserted into E. coli.
Once inside a cell, the bacterial machinery treats the AI-generated DNA sequence as if it were a normal phage genome, making viral proteins and constructing new phages from scratch. If a design works, the phages will infect and kill their host bacterium, and then show up as clear spots on agar plates. Just 16 of the AI-generated genomes “booted up” to form viable bacteriophages.6
In one experiment, the researchers mixed all 16 AI-generated phages with ΦX174 and then threw them into a tube with E. coli cells. Because the phages were forced to compete for the same host cells, the variants that reproduced fastest would dominate. By sequencing the phages over time, the researchers could track which phages were gaining ground and which were falling behind. Several of the AI phages consistently outperformed wild ΦX174, with one variant (called Evo-Φ69) increasing to 65-times its starting level.
Ultimately, these 16 AI-generated phages were not only viable; in many cases, they were more infectious than wildtype ΦX174 despite carrying major genome alterations that a human would be unlikely to rationally design.
Consider Evo-Φ36, an AI-designed phage with eleven genes, just like wild ΦX174. One of this phage’s genes, called J, was swapped with a gene from a distant phage, called G4. But the J protein from phage G4 is much shorter than the J protein in ΦX174, with past experiments showing that this swap cripples the phage. And yet, somehow, the AI model rewired the rest of the Evo-Φ36 genome to make this swap work, thus producing a viable and infectious virus. This kind of context-dependent compatibility is hard for human designers to anticipate, but it emerges naturally from AI models that can integrate patterns from thousands of related genomes.
Genome Design at Scale
A theme at the heart of synthetic biology is that well-characterized DNA sequences can be joined together to build new, desirable functions in living cells.
This ideology has worked remarkably well for “toy” systems, such as toggle switches, oscillators, and a few proof-of-concept therapies. But it does not easily scale to larger systems. Cells are not modular gadgets, but instead have layer upon layer of feedback loops and emergent properties. Even with massively parallel experiments, we likely cannot brute-force our way to new organisms by modeling every cog and gear.
Perhaps this is why this new preprint feels so compelling. Rather than designing genomes piece by piece and troubleshooting each failure, this study demonstrates that we can leverage data and better DNA synthesis methods to more efficiently explore biological “space.” And, further, that we can use those methods to build viable organisms that significantly deviate from natural versions.
Several of the viable AI phages reported in this paper, for example, shared less than 95 percent of their genome with any known phages. In evolutionary terms, this would denote them as a new species.7
Of course, this paper will probably be alarming to folks in the biosecurity community. The authors point out that Evo 2 excludes human viruses from its pretraining data, and also show that "both pretraining and fine-tuning [are] necessary for coherent generation of complex systems." Since the Evo 2 model is fully open, though — including its "training code, inference code, model parameters, and…training data” — then a sufficiently motivated person could, in principle, sculpt these models to design human viruses. The HIV genome is only about 10,000 bases in length (not much larger than bacteriophages) and the coronavirus genome is about 30,000 bases.
Although the risk of training on human viruses seems troubling, the real barriers to moving from phages to larger organisms are data and atoms.
Even a simple bacterium like E. coli has a genome three orders-of-magnitude larger than ΦX174. Using these models to design bacteria or human cells, then, will demand far more curated datasets and way more compute.
Even then, there is still the issue of DNA synthesis and assembly. Building hundreds of small phage genomes to test in the laboratory is not too expensive, but building hundreds of bacterial genomes would put most biotechnology companies out of business. In other words, using AI to make synthetic genomes costs a great deal at both the design and construction stages.
For context, Jason Chin’s group at the University of Cambridge recently designed and chemically-synthesized a full E. coli genome with about four million bases of DNA. This E. coli genome was engineered to contain only 57 codons, instead of the normal 64. Some of the codons that cells use to build proteins are redundant — CUU, CUC, CUA, CUG, UUA, and UUG all encode the amino acid leucine, for example — and so can be removed and replaced with codons for “noncanonical” amino acids. These “redesigned” cells can thus be coaxed to build proteins containing a broader palette of amino acids. But this genome synthesis effort took several years of work and reportedly cost more than a million dollars.8
It’s also worth asking whether this kind of full-genome design is as useful for bioengineering as it initially seems. In many cases, especially with viruses, a handful of genes drive the traits that matter most. Adeno-associated viruses, for example, infect specific tissues in the human body due to particular proteins embedded in their capsids. By tinkering with the genes encoding those proteins, one can alter which cells these viruses infect. And so, with this logic in mind, perhaps the wholesale, bottom-up design of an entire phage genome may be more of a technical milestone than a practical one. Using AI to design single genes or regulatory regions — where you know which knobs you’re turning — may, in many cases, be the more efficient strategy.
And finally, although the fine-tuned model in this paper designed viable phages with hundreds of novel mutations, the actual biology of those phages did not change — they still infect E. coli, just as ΦX174 does. In synthetic biology, though, we often care about creating new functions. The next step, it seems, will be to design some sort of “training feedback loop” with which these AI models can be coaxed to build phages with pre-specified behaviors.
I’m quite confident this will happen soon. When Frederick Sanger published the first full genome sequence of ΦX174 in 1977, using a DNA sequencing technology called di-deoxy termination, the final sequence was only 5,375 bases long. At the time, “that number represented the outer limit of gene-sequencing capability,” Siddhartha Mukherjee wrote in his 2016 book, The Gene: An Intimate History. Scaling it up to the human genome, with its 3 billion base pairs, represented “a scale shift of 574,000-fold.” And yet it happened within a quarter-century, using technologies that were not all that different from Sanger’s original method.9
In this light, this new paper feels a bit like history repeating itself. Just as Sanger’s sequencing of ΦX174 opened the path toward modern genomics, these AI-designed phages may indeed mark an early step in the new field of "Genome Design." And as history shows, the leap from 5,000 bases to 3 billion may not be as insurmountable as it seems.
Niko McCarty is a founding editor of Asimov Press.
Cite: McCarty, Niko. “AI-Designed Phages.” Asimov Press (2025). https://doi.org/10.62211/21er-45fg
Thanks to Eryney Marrogi and Alec Nielsen for reading drafts of this. Lead image by Ella Watkins-Dulaney.
In the 1950s, virologists used the Greek letter phi (Φ) to denote bacteriophages as a distinct type of virus that infects specifically bacteria.
Even today, the ΦX174 genome is used as a reference or “standard” control on DNA sequencing machines.
The length of this “seed” prompt is extremely important. From the paper: “We found that prompting with the first nine or more nucleotides from the ΦX174 consensus sequence led to simple, memorized recall of ΦX174 with minimal diversity.” But “prompting with only the first one or two nucleotides from the consensus sequence did not provide sufficiently strong conditioning to produce ΦX174-like generations.” The sweet spot, then, seems to be about 4-8 nucleotides.
It likely carried a mutation that rendered one of the phage proteins nonviable.
The 17 “failed” genomes may have contained long, repetitive regions, which are difficult to stitch together.
From the paper: “When we compared the generated genomes to Microviridae sequences in the training data, they contained between 67 and 392 novel mutations, with nucleotide sequence identities between 93.0% and 98.8%.”
In the thrilling world of phage taxonomy, phage genomes that share less than ~95 percent average nucleotide identity with known phages are typically classified as a new species. At least one of these AI-designed phages, Evo-Φ2147, would meet that threshold.
Some public data: It costs about $0.07 to order one base of DNA from Twist Biosciences, and ordering large or “difficult” sequences (such as from GenScript) usually drives that price up closer to $0.45 per base because companies need not only to chemically synthesize short strands of DNA but then stitch them together.
In Sanger’s original chain-termination method, DNA was copied in the presence of “normal” nucleotides and also a small concentration of “dideoxynucleotides.” These other nucleotides lack the 3′ hydroxyl group that DNA polymerase needs to extend a chain, so whenever one is added, the chain stops growing. If you copy DNA in the presence of these dideoxynucleotides, then they will randomly get incorporated while the DNA strands are at unique lengths. And then, by running four parallel reactions — each spiked with a different dideoxy base (A, T, G, or C) — you get a collection of fragments ending at every possible position of that base. These fragments are then separated by size on a gel, allowing the DNA sequence to be read directly from the pattern of bands. Later, chemists added fluorescent dyes to each dideoxynucleotide so that all four reactions could be combined and run in a single lane, with the sequences read automatically by a detector.
 | A guest post by
|
Really nice post, Niko, and a pretty wild result. After the Evo 2 paper, I thought viable genome generation was still further out. However, I think you're glossing over the downsides a tad too quickly.
You correctly point out that this result has biosecurity folks worried since Evo 2 is an open-weights model and many viruses of concern are only a few kb longer than ΦX174.
But your bottom line from that is, to me, a little bit disconnected: "Although the risk of training on human viruses seems troubling, the real barriers to moving from phages to larger organisms are data and atoms."
It is no doubt correct that larger organisms are much much more challenging to generate and synthesize, but that has little to do with the aforementioned worries about human viruses—and I do worry a lot at this point!
I think that a method that was able to de novo generate very sequence-divergent, viable viruses warrants a pretty thorough dual-use discussion, especially after we've seen what a little bit of genetic distance and a few kb more did in late 2019...
"Of course, this paper will probably be alarming to folks in the biosecurity community. The authors point out that Evo 2 excludes human viruses from its pretraining data"
This is unfortunately not entirely true, at least based on my reading of the methods section of the Evo 2; they *tried* to exclude human viruses but in fact I believe the training set included a large amount of viral sequence. This new result is quite impressive but I really wish they had consulted a bit more widely before releasing it. I don't think at this point it is an immediate biosecurity threat but (as someone who is typically fairly dismissive of AI biosecurity concerns) this paper is genuinely concerning to me.