
A Visual Guide to Gene Delivery
There are at least 10,000 known monogenic diseases. When attempting to cure them, how do clinicians decide which gene-therapy delivery method is best?
A gene is the essential hereditary unit that passes traits from parents to offspring; a segment of DNA containing instructions for making a specific protein or molecule that performs a particular function in your body. And yet, despite being fundamental to our flourishing, genes can be broken.
Researchers have identified nearly 10,000 monogenic diseases, or conditions caused by errors in a single gene. Examples include hemophilia, which once plagued the royal houses of Europe, and sickle cell anemia, which affects around 8 million people worldwide.
Fortunately, as readily as these diseases are discovered and characterized, so too are many of their cures. As methods for resolving errors in DNA continue to advance, it has never been easier to fix a broken gene. Just this year, a baby with CPS1, a disease that causes ammonia to accumulate in the blood, became the first recipient of a CRISPR-tailored gene cocktail. The gene therapy is designed to protect this infant from the neurological disorders that otherwise would have resulted from this rare, and often fatal, condition. And while matching a gene therapy to its targeted mutation is an intricate and nuanced process, much boils down to two core elements: a vector and its cargo.
The first of these components is the therapeutic genetic payload, or cargo, which is designed to correct or regulate the disease-causing mutation, whether that means offering a healthy copy of a mutated gene or delivering precision-editing tools like CRISPR. Equally vital is the delivery vector, which carries this payload to specific cells.
There are, broadly, two types of gene therapy: ex vivo and in vivo. For the former, clinicians extract a patient’s cells, engineer them in a laboratory, and then infuse them back into the body. CAR-T therapy (perhaps the most promising cancer treatment of the last several decades) is one example, in which scientists extract a patient’s blood cells, modify T cells so they can recognize and attack malignancies more effectively, and reinfuse them back into the body. Casgevy, the first FDA-approved CRISPR-based therapy, is also an ex vivo gene therapy; it aims to cure sickle cell disease by editing a patient’s blood-producing stem cells so they generate functional fetal hemoglobin, then returning these corrected cells to the bloodstream.
Many disorders, though, can only be cured using in vivo gene therapies delivered directly into the body. Researchers cannot extract, edit, and replace neurons the way they do blood cells, so diseases involving the brain, muscles, or liver require vectors that reach and fix cells in their natural environments. There are many in vivo gene therapies under development for conditions like Duchenne muscular dystrophy, spinal muscular atrophy, and von Gierke’s disease.
While the nature of the therapy and the target cell type determine which of these methods is used, selecting the delivery vehicle for a gene therapy basically comes down to balancing tradeoffs. Some diseases occur so rarely that they face treatment barriers due to unfavorable economics of scale. Many monogenic diseases appear at rates of 1 in 100,000 on the higher end. Companies weigh market size, reimbursement potential (the likelihood insurers or governments will pay for the therapy), and the level of existing competition before deciding which vector to use for their gene therapy. Manufacturing advanced vectors at scale can drive prices for these treatments into the millions. Thus, the core mechanism driving the disease may be well-established and thus easy to target, even as funding for research and development is difficult to procure.
And even if all these economic hurdles are overcome, there are still three main technical challenges to solve: cargo capacity, specificity, and immune response.
First, cargo capacity (the maximum size of the genetic payload) limits whether a particular vector can be used. Some diseases are caused by mutations in large genes that exceed the packaging limits of existing vectors like adeno-associated virus (AAV). Duchenne muscular dystrophy, for example, is caused by mutations in the dystrophin gene, which spans roughly 14 kilobases (kb) — far larger than the ~4.7kb capacity of natural AAV vectors.
Second, each vector has tissue specificity, meaning certain strains (like AAV9) target neurons better, while others (such as AAV8) home in on the liver. This preference determines how well a therapy reaches its intended site without straying into non-targeted tissues. Vectors also differ in the duration of their expression. In other words, once a virus or non-viral vector finds the target cell and releases the cargo, that cargo can either stick around for a while by integrating into the host’s DNA or it can clear out quickly.
And lastly, viral vectors can be immunogenic, meaning they trigger an immune reaction. The human immune system is designed to recognize and eliminate foreign material, which unfortunately includes common viral vectors for gene therapy. Certain organs, such as the liver, are particularly immunologically active and may mount stronger immune responses against gene therapy vectors. In contrast, the eye, with its lower immune activity, offers a more receptive target.
Taken together, these technical and economic considerations complicate which delivery method is best for gene therapies. Indeed, “best” makes little sense outside the context of specific therapies. To better understand the various tradeoffs at work, then, let’s examine the various gene delivery vehicles both on the market and under development.
This article includes a poster. You can download a free PDF version or buy a framed copy in our Asimov Press store.

Adeno-Associated Virus (AAV)

Whereas some of the first gene therapies made use of retroviruses, which replicate by integrating their genetic material into the host cells’ DNA, those therapies often triggered harmful immune responses. This changed with the discovery of AAVs.
First identified in 1965, AAVs are small, replication-deficient viruses that rarely cause disease in humans and offer a safer, more predictable alternative for delivering therapeutic genes.
Robert Atchison and his colleagues at the University of Pittsburgh discovered AAVs while sifting through unusual viral particles found in lab cultures that had been infected with adenovirus. Atchison was surprised to find that these smaller particles could not reproduce on their own, and that the adenoviruses themselves supplied crucial proteins for their replication.
Though many know it today in the context of gene therapy, AAV’s potential as a vector didn’t emerge until 1995, when it was first tested in a patient. That year, Terrence Flotte’s team at Johns Hopkins University used a natural strain of AAV, called AAV2, to introduce a cystic fibrosis transmembrane conductance regulator (CFTR) gene — defective in those with cystic fibrosis — into patients during a phase 1 clinical trial. Although the therapy did coax cells to make functional CFTR proteins, patients showed only modest clinical improvements.
AAV is a small virus that lacks a lipid membrane and carries a single-stranded piece of DNA stretching 4.7 kb. Its exact packaging size can vary by serotype, and there are now more than 20 antigenically distinct types of AAV. These different serotypes follow a loosely standardized naming system, commonly AAV-X, where X stands in for a number indicating a particular variant. Different serotypes tend to target certain cell types, and before AAV engineering became common, this natural “tropism” determined their application.
When AAV infects a cell, it first latches onto cell surface receptor proteins. The specific surface receptors differ between serotypes. The AAV then induces endocytosis, a mechanism where the cell engulfs external particles by folding them into membrane-bound vesicles. After internalization, AAV escapes from endosomes and moves to the nucleus. Once inside, its single-stranded DNA converts to double-stranded DNA, which persists in the nucleus but does not typically integrate into the host genome.
The DNA delivered by AAV typically wraps up into a circle, or mini-chromosome, called an episome. Although AAV DNA usually remains outside the cell’s chromosomes, evidence suggests integration sometimes happens, especially when DNA double-strand breaks occur, challenging the traditional notion of AAV as a purely non-integrating vector. These episomes can persist in non-dividing cells such as neurons or cardiac myocytes for years. But in rapidly dividing cells, they become diluted over multiple rounds of replication, leading to the need for repeated dosing if long-term therapeutic benefit is required.
Traditionally, researchers chose a serotype based on which organ they aimed to repair, then administer the therapy by the route best suited for that tissue: intravenous (IV) for systemic delivery, intramuscular (IM) for muscle or lymphatic targets, direct injection into the eye or spinal cord, or, in the case of lung disease, via a nebulizer (a medical device that turns liquid medication into an inhalable mist). AAV1 and AAV6 often target the liver effectively, for example, while AAV9 can cross the blood-brain barrier and deliver gene therapies into the central nervous system.
A few companies, including Dyno Therapeutics, are engineering viral capsids capable of targeting and entering specific cell types. Dyno’s approach draws on AI-designed libraries of millions of engineered viruses, which they produce and test in non-human primates. The company has had success in designing brain-specific variants showing 50- to 100-fold improvements over natural AAV9, as well as eye-directed variants that outperform standard capsids by a wide margin. Capsida Therapeutics employs a similar AI-guided direction to capsid design, while Voyager Therapeutics’ TRACER platform, which relies on traditional logic-driven R&D, has led to best-in-class blood-brain-barrier penetrant AAV capsids.
Each of these companies aims to modify the cap gene through some type of evolution. When it comes to AI-design, this is directed evolution, where diversity is generated at scale with the hope that eventually new capsids emerge that can evade natural neutralizing antibodies (NAbs), create tissue-specific tropism, and, in some cases, increase packaging size.
A continuing challenge for AAV therapies is how to get them into the body without triggering an immune response, which are prone to happen with large initial dosing common to systemic therapies. Pre-existing NAbs — developed from natural AAV exposure — can block transduction by attaching to incoming viral particles. Studies suggest that in certain populations, up to 80 percent of individuals may carry circulating NAbs against specific AAV serotypes, posing major hurdles to therapy. Even in patients without NAbs, the humoral immune system can target transduced cells via CD8+ T-cells, which eliminates those cells before the therapy takes effect. This phenomenon may affect as many as 50 percent of candidates.
Though AAV’s immune profile is rather mild in most patients, deadly complications can still arise with this vector. Because of innate immune recognition of injected AAVs, patients in certain trials have experienced liver failure, thrombotic microangiopathy (a disorder causing abnormal blood clots in small vessels), or other organ failures. One recent Duchenne muscular dystrophy trial saw such outcomes at high doses, illustrating the trade-off between sufficient gene delivery and immune risk.
AAV tops our list because this vector has successfully shuttled several FDA-approved treatments. Luxturna (AAV2 expressing the human retinal pigment epithelium 65 kDa protein to correct retinal dystrophy), Zolgensma (AAV9 vector for spinal muscular atrophy), and Hemgenix (AAV5 vector for hemophilia B).
Still, AAV’s most obvious limitation remains its packaging capacity (~4.7 kb). This initially posed challenges for delivering the large Cas9 enzyme (which is encoded by a 4.2kb gene) plus its guide RNAs and necessary control elements. Researchers have met this constraint by using smaller Cas9 orthologs, such as SaCas9 from Staphylococcus aureus (~3.3kb), by splitting SpCas9 across two AAV vectors that reconstitute in the host, or by employing a dual-vector strategy where one AAV delivers Cas9 and another provides guide RNAs. No one has yet made or discovered a capsid that optimally combines precision targeting of a cell type with a large enough packaging capacity to resolve all these problems.
Adenovirus

Adenoviruses were discovered in 1953 by Wallace Rowe, who isolated them while investigating a respiratory illness in tissue derived from “adenoid” cultures (cells taken from tonsils in the back of the throat). Though they share a similar name with AAV, adenoviruses are distinct: they are medium-sized, about 90 nanometers in diameter, and non-enveloped, carrying a double-stranded DNA genome of about 36 kb — significantly larger than AAV’s capacity of around 4.7 kb.
In 1993, Ronald Crystal’s team at Weill Cornell used a genetically altered cold adenovirus vector in an early gene therapy trial for cystic fibrosis, hoping to restore CFTR function in patients with the disease. Unlike the 1995 AAV-based study, this trial exposed serious limitations of using adenoviruses in gene therapy, as participants mounted strong immune responses. One particularly severe outcome contributed to what many describe as a “gene therapy winter.” In 1999, Jesse Gelsinger, a participant in a trial at the University of Pennsylvania to treat his inherited ornithine transcarbamylase deficiency, received an adenovirus-based therapy but died four days later following a massive immune reaction.
Adenovirus infects cells by attaching to surface receptors (such as coxsackievirus and adenovirus receptor, or CAR) and getting pulled inside. Once there, it escapes its membranous container and travels to the nucleus, where it releases its DNA. The viral DNA then remains separate from the cell’s chromosomes (as episomes) rather than integrating into them. Both adenoviruses and AAVs primarily persist this way, though AAVs can occasionally integrate into the host genome while adenoviruses rarely do.
However, adenoviral genomes tend to be lost in cells that divide frequently, both because the immune system clears the virus and because the viral DNA does not integrate. As a result, adenoviruses deliver more transient gene expression compared to other vectors. In some settings, this can be beneficial — for example, in cancer therapies or any situation where short-term expression is sufficient — but it can be limiting in conditions requiring long-term correction.
Adenoviruses have nearly 100 identified serotypes (classified into distinct camps, ranging from species A through G), and each has distinct tissue preferences determined by its capsid proteins. Species C, which includes Ad1, 2, 5, and 6, often targets the respiratory tract and is one of the most commonly used in gene therapy studies. Species B (Ad3, 7, 11, and 14) tends to focus on the kidney and urinary tract. Adenoviruses have a natural tropism for the liver and spleen, which can hamper efforts to treat other organs.
A major drawback of adenoviruses involves immunogenicity. Common serotypes such as Ad2 or Ad5 frequently trigger strong innate and adaptive immune responses, activating pattern recognition receptors like TLR9 (which detects viral DNA) and TLR2 (which recognizes capsid proteins). This stimulation causes the body to make inflammatory cytokines such as IL-6, TNF-α, and type I interferons. Neutralizing antibodies often form and prevent effective re-administration, while CD8+ T-cells can attack transduced cells and shorten the duration of transgene expression.
Pre-existing immunity is also problematic, as up to 70 percent of adults may carry antibodies that inactivate common human adenoviruses before they can work. This hurdle has sparked efforts to develop rare human serotypes, non-human adenoviruses (for example, chimpanzee adenovirus), and utilization of natural chimeric viruses. A naturally-occurring “human-chimp” capsid was discovered in Sweden by “adeno hunters,” who search for novel adenovirus variants in the wild rather than engineering them in the lab. These natural chimeras present an evolutionary solution to the problem of immune evasion currently addressed through engineering. Unfortunately, the need for multiple doses in some applications increases the chance of severe immune reactions, as in Jesse Gelsinger’s tragic case.
Still, when carefully managed at appropriate doses, adenovirus shows significant promise. It serves as the backbone for vaccines that have been deployed worldwide to prevent COVID-19 (Janssen, Vaxzevria, Sputnik V) and Ebola virus. Beyond infectious disease prevention, adenovirus also exhibits strong potential in cancer therapy. The FDA recently approved an adenoviral vector-based gene therapy for high-risk, unresponsive non-muscle invasive bladder cancer, and additional oncology applications are under investigation. Advexin, a therapy for advanced recurrent head and neck squamous cell carcinoma, is currently in phase III trials.
Like AAV, the future of adenovirus in gene delivery depends on capsid engineering and the construction of hybrid vectors that merge components of different viruses. Researchers hope these innovations will produce safer, more effective therapies that can overcome the obstacles of pre-existing immunity, strong immune responses, and the limitations of transient gene expression. After the Gelsinger case, work on this vector has lagged behind AAV. So even though capsid engineering is on the horizon for adenovirus, it has some catching up to do.
Lipid Nanoparticles (LNPs)
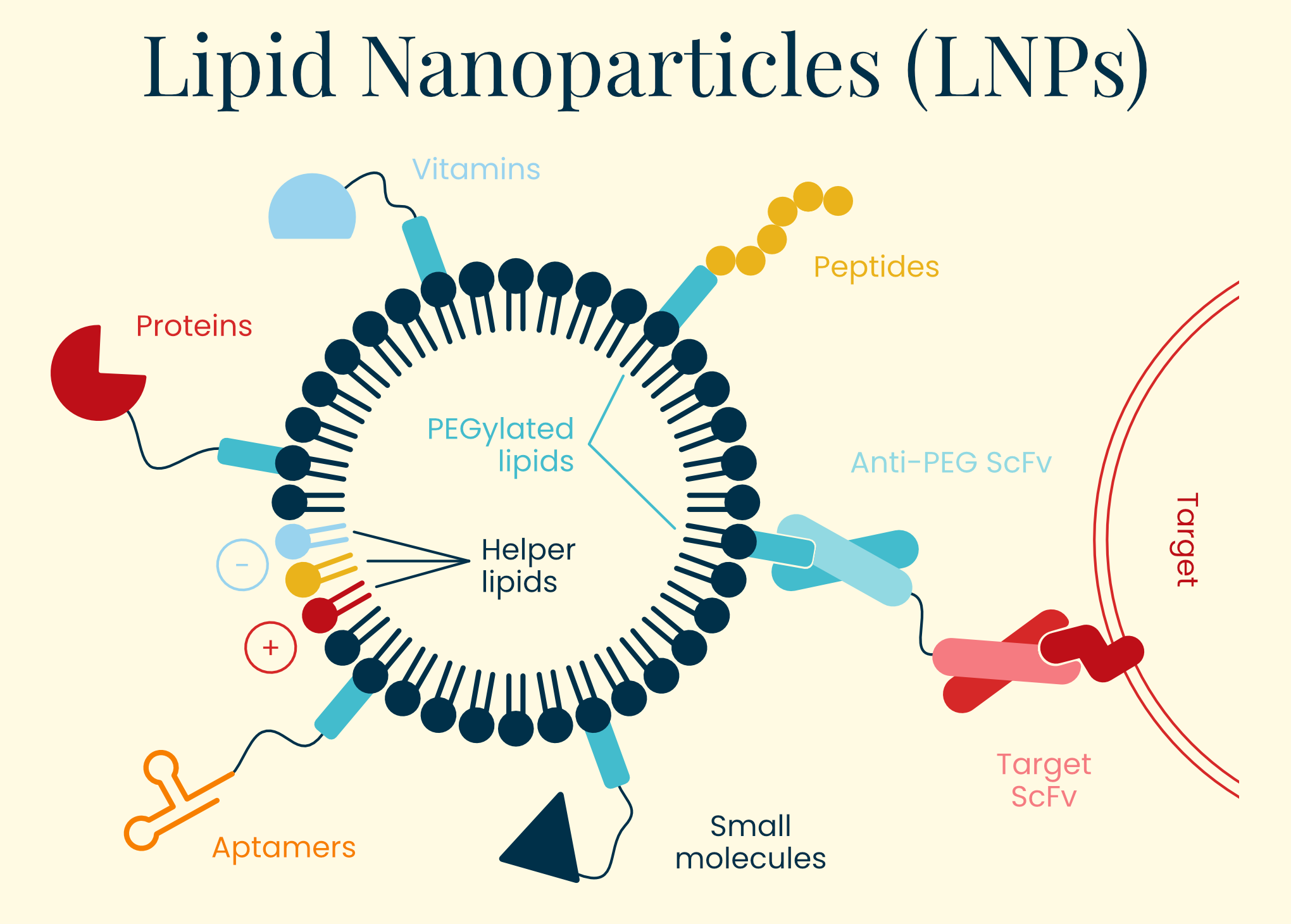
Lipid nanoparticles (LNPs) are microscopic spherical structures made of fat-like molecules that encapsulate and protect nucleic acids like mRNA for delivery into cells. As delivery vehicles, they trace their roots to 1960s liposome research, evolving into sophisticated systems during the 1990s and 2000s. Early RNA therapies faced rapid degradation, immune recognition, and poor cellular uptake until Katalin Karikó and Drew Weissman showed that modifying certain nucleosides in mRNA (replacing uridine with pseudouridine) could reduce unwanted immune responses while preserving protein production — an achievement that earned them the 2023 Nobel Prize in Medicine.
LNPs measure between 50 and 120 nanometers and primarily carry functional RNA — such as mRNA or guide RNAs for CRISPR — rather than DNA. This payload directs host cells to produce a therapeutic protein within hours of successful delivery, and because RNA is relatively compact, scientists can fit multiple mRNAs into a single LNP. Lipids form stable complexes that shield the genetic material from degradation during therapy manufacture. Once injected, a PEG-lipid coating helps LNPs avoid clumping and reduces rapid clearance by immune cells, although this protective layer often sheds within minutes.
After LNP absorption into the targeted cells, their protective shell breaks down, releasing their nucleic acid cargo into the cytoplasm. This process works perfectly for mRNA-based therapies since mRNA only needs to reach the cytoplasm to be translated into proteins by the cell's ribosomes. For gene editing applications, LNPs can deliver mRNA encoding editing proteins (like Cas9) along with guide RNAs. Once inside the cell, these mRNAs are translated into proteins that can then enter the nucleus to modify DNA.
Historically, LNPs excelled at three main targets: muscle and lymph nodes when injected intramuscularly, the lungs via inhalation, and the liver when delivered intravenously. Without targeting modifications, intravenous LNPs accumulate in the liver, making them ideal for clotting factor diseases like hemophilia, but less efficient for other tissues. When employed in vaccines, LNPs enter the muscle tissue, where they transfect local muscle and immune cells. Any systemic distribution to the liver is generally an unintended detour, presenting a considerable hurdle for any LNP therapy trying to avoid getting trapped in the liver.
Recent engineering advances have significantly broadened LNPs’ reach through several approaches. Helper lipid modification can redirect them by altering phospholipid charge — neutral helpers favor liver accumulation, positive charges enhance lung targeting, and negative charges direct toward the spleen. Small molecule conjugation offers another approach, with molecules like GalNAc enhancing hepatocyte targeting through asialoglycoprotein receptors (useful for patients lacking LDLR), vitamin A derivatives targeting cells involved in fibrosis, and bisphosphonates targeting bone tissue.
Perhaps most promising is antibody conjugation, which enables highly selective targeting to specific cell types. Anti-CD3 or anti-CD5 antibodies direct LNPs to T cells (enabling in vivo CAR-T therapy), while anti-CD117 targets hematopoietic stem cells — potentially allowing less invasive treatments for blood disorders like sickle cell disease. This approach presents the possibility of directing LNPs to virtually any blood or immune cell type of interest, resolving the limited distribution problem that has limited LNPs to this point.
Beyond vaccines — like Moderna’s and Pfizer-BioNTech’s COVID-19 products — LNPs have advanced gene editor delivery. In 2015, the FDA approved Onpattro, an LNP-based siRNA therapy for transthyretin amyloidosis. And as mentioned earlier, in May of this year, researchers at the University of Pennsylvania made headlines by custom-designing an N-of-1 LNP-based gene therapy for a baby. In the Penn study, the patient actually safely received two doses – a low then a higher one – of the therapy without major adverse effects, spaced a few weeks apart. Clearly, this vector works.
Although no commercial products yet combine siRNA and DNA in one package, Roche’s acquisition of Poseida Therapeutics suggests continued interest in LNP-based DNA delivery. Meanwhile, Beam Therapeutics is developing LNP-based therapies for alpha-1 antitrypsin deficiency and glycogen storage disease, along with potential autoimmune treatments. Moderna is also pursuing additional LNP-enabled vaccines that aim to replicate the success of its COVID-19 platform.
Even though LNPs are non-viral and so less likely to provoke strong, systemic immune reactions, challenges remain. Scientists are testing new ionizable lipids that minimize inflammation, adding immunosuppressive agents such as dexamethasone, and further refining mRNA design (for example, by substituting naturally occurring nucleosides or purifying the final product via high-performance liquid chromatography). These tweaks can reduce innate immune detection and allow longer therapeutic activity.
An advantage LNPs have over viral vectors hinges on how effectively they deliver mRNA, giving rapid action ideal for particular cell types. Fast-dividing cells, like those in the skin and lung epithelium, are prime targets for LNPs, where cheap, recurrent treatments are viable. Even with these upsides, making LNPs work for gene therapy will require improvements in both targeting and avoiding immune responses.
The greatest advantage in the use of LNPs lies in its affordability. The total production costs, including raw materials and labor, for the Pfizer-BioNTech and Moderna mRNA vaccines cost just $1-3 per dose. Coupled with greater ease of redosing, again demonstrated most recently in Penn’s N-of-1 individualized gene therapy in 2025, LNPs are effective for liver targeting where redosing might be required. Even though gene therapy applications might require larger and regular doses, the charge per dose would not prohibitively increase.
Herpes Simplex Virus (HSV)

Gene therapy isn’t generally the first thing people think of when herpes is mentioned, but it’s now being used as a viable delivery vector. Herpes simplex virus (HSV) has a long history of documentation in human disease, first identified in ancient Greek writings as "herpes" (meaning to creep or crawl). Herpes’ infectious nature wasn't experimentally confirmed until 1919, when German physician A. Lowenstein successfully infected rabbits with the virus. This family of viruses is ubiquitous in the human population. HSV-1 (oral herpes), for example, is present in about 67 percent of people under age 50 worldwide and HSV-2 (genital herpes) affects approximately 11 percent of people.
What makes HSV particularly interesting for gene therapy applications is its natural neurotropism, or the ability to infect and establish long-term latency in neurons. HSV travels up nerve endings to neuronal cell bodies where it establishes latency (dormancy), but can periodically reactivate and travel back down the same nerve pathway to cause recurrent symptoms at the original infection site. This explains why cold sores appear repeatedly in the same location on the mouth.
Additionally, HSV has an exceptionally large genome (~152 kbp) compared to other viral vectors like AAV (~4.7 kb), allowing it to carry substantial genetic payloads for therapy. These features make HSV an enticing option in combinatorial therapies for well-characterized complex diseases involving multiple genes.
The herpes life cycle is well-characterized. HSV enters cells by binding to cell surface receptors, primarily through interactions of viral glycoproteins with heparan sulfate proteoglycans, followed by fusion with the cell membrane. Once inside, HSV can either enter a lytic cycle (actively replicating and killing the host cell) or establish latency, particularly in neurons. This dual lifecycle provides unique opportunities for gene therapy applications, as the virus can be engineered to either express therapeutic genes transiently or maintain long-term expression in non-dividing cells.
The breakthrough application of HSV in gene therapy came with Talimogene laherparepvec (T-VEC, marketed as Imlygic), which received FDA approval in 2015 as the first oncolytic virus therapy for advanced melanoma. T-VEC is derived from HSV-1, genetically modified to selectively replicate in tumor cells while sparing normal tissue. Two key modifications drive its specificity: deletion of the ICP34.5 gene (reducing neurovirulence and allowing preferential replication in cancer cells with defective antiviral responses) and deletion of ICP47 (which normally blocks antigen presentation, but its removal enhances anti-tumor immune responses). In other words, this therapy works by repurposing the features that make HSV a stubborn disease-causing virus in humans to kill tumor cells.
T-VEC carries an inserted gene for granulocyte-macrophage colony-stimulating factor (GM-CSF), which helps recruit and activate immune cells to the tumor site. When injected directly into melanoma lesions, T-VEC infects and lyses cancer cells, releasing tumor antigens and GM-CSF into the tumor microenvironment. This creates a personalized, in situ cancer vaccine effect, potentially generating systemic immune responses against both injected and distant tumors.
Given their neuron-seeking behavior and demonstrated successes in cancer, HSV presents a unique option for the future of glioblastoma. Several trials, notably the 2021 trial of G207 for pediatric high-grade gliomas, are using oncolytic HSV-1 mutants with deletions in both copies similar to T-VEC to restrict tumor replication.
Another unique aspect of HSV biology is its ability to be transported along axons in both anterograde and retrograde directions. This enables vector delivery to specific neuronal populations by peripheral injection, avoiding invasive brain procedures. For instance, HSV vectors injected into the skin can travel retrograde along sensory neurons to deliver therapeutic genes to dorsal root ganglia for treating neuropathic pain.
As with all vectors, the immune response remains a challenge for HSV vectors, as pre-existing immunity to HSV is common in the human population. That is to say, if someone has previous exposure to HSV (upwards of 63 percent of American adults do), it is not a safe option. Additionally, HSV has inherent cytotoxicity, even in its engineered forms, which can limit long-term gene expression in some applications. Research continues on reducing the immunogenicity and cytotoxicity of HSV while maintaining its gene delivery capabilities.
Lentivirus
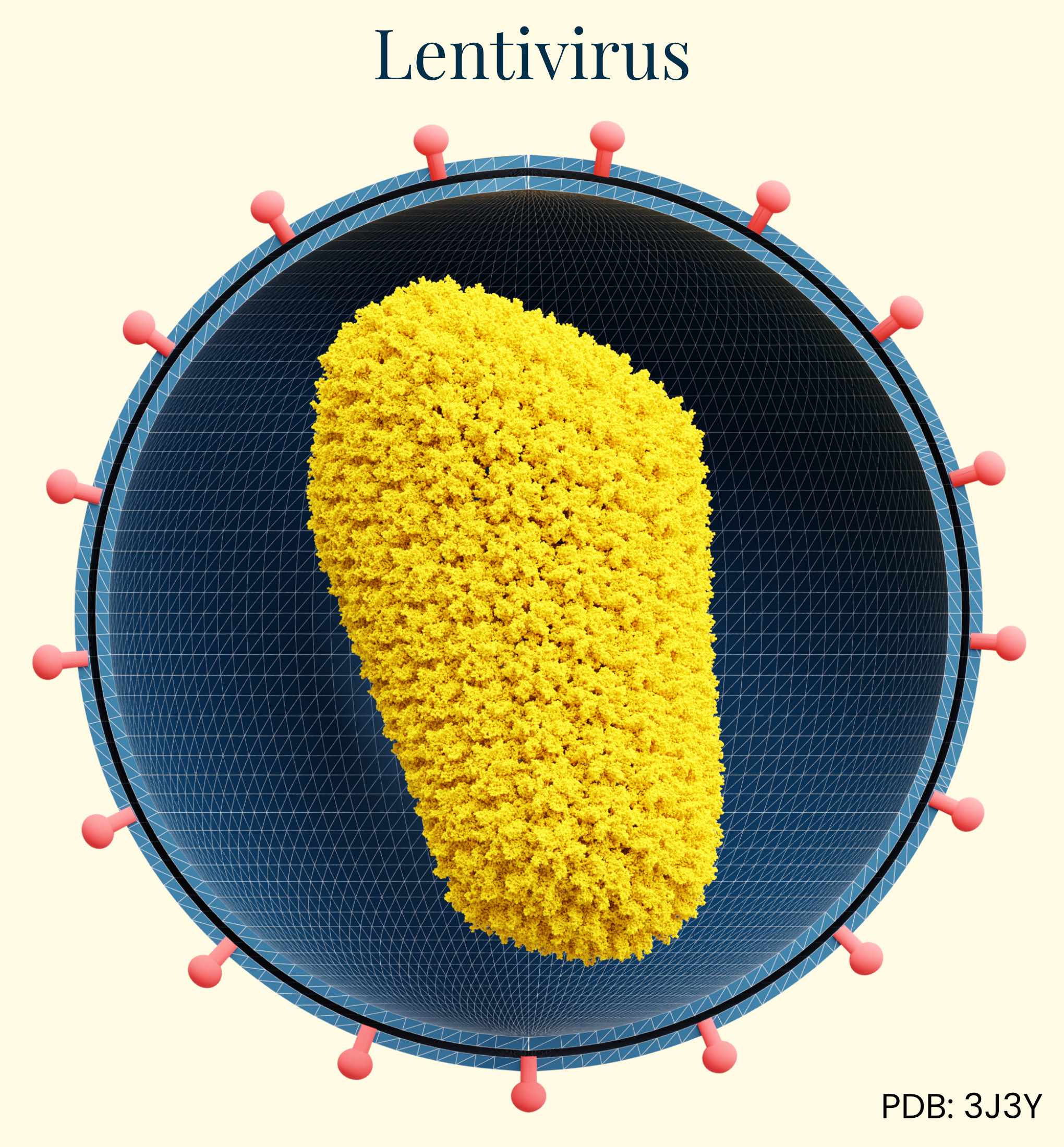
Our final class of vectors, lentiviral vectors derived primarily from HIV-1, are enveloped retroviruses with an RNA genome of approximately 9kb. During the 1990s, researchers recognized the potential of a modified, non-pathogenic form of lentivirus as gene therapy vectors. Able to integrate genetic material into non-dividing cells, they addressed a major limitation of earlier retroviral vectors. Unlike AAV and adenovirus, lentiviruses integrate their genetic material into the host genome, enabling permanent modification and persistent transgene expression in both dividing and non-dividing cells.
The delivery mechanism begins with pseudotyped envelope proteins (commonly VSV-G) binding to cell surface receptors, followed by endocytosis, reverse transcription of the viral RNA genome into DNA, nuclear entry, and integration into the host chromosome. This integration capability makes lentiviral vectors particularly valuable for ex vivo cell therapies where cells are removed from a human, edited, and then returned to the body. However, it raises concerns about insertional mutagenesis for in vivo applications. Because the genome integrates into the host cell, insertional mutagenesis can disrupt normal gene functions or even activate oncogenes, creating concern for swapping one disease for another, later on.
Lentiviral vectors show broad tropism that can be modified through pseudotyping with various envelope proteins. Their immunogenicity profile is moderate compared to adenovirus, with primary responses directed against the transduced cells rather than the vector itself.
The most successful clinical applications have been in ex vivo gene therapies, including FDA-approved CAR-T cell cancer therapies (Kymriah, Yescarta) and treatments for primary immunodeficiencies, β-thalassemia (Zynteglo), and metachromatic leukodystrophy (Libmeldy). No market in vivo applications exist, due in part to the manufacturing scalability, stability, and insertional mutagenesis risks, which have limited their advancement compared to non-integrating vectors like LNPs, AAV, or even adenovirus.
Finding the Right Tool
Every vector system — viral or otherwise — has tradeoffs regarding duration of expression, specificity, and payload capacity. Diseases requiring long-term expression will demand different therapies than those requiring only transient effects, as in correcting liver abnormalities. A highly specific cell type target will require a different choice than a general region. Construct size also determines vector selection. AAV demonstrates excellent safety profiles and persistence in non-dividing cells, and capsid engineering efforts improve the direction of tropism, but payload capacity remains a concern. LNPs excel in delivering mRNA for transient expression that can be dosed repeatedly, but directed tissue targeting has thus eluded researchers. Lentivirus is an excellent choice for ex vivo applications.
To grasp these tradeoffs even more concretely, let’s consider how they coalesce in the context of a speculative gene therapy. Back in February, I wrote about DEC2 (BHLHE41), a gene encoding a basic helix-loop-helix transcription factor that regulates circadian rhythm and sleep homeostasis. Specific mutations in DEC2 drive changes in sleep needs by making sleep more efficient (at least in mice). So let’s say we wanted to design a gene therapy to introduce the short sleeper genotype, DEC2, into humans. Which delivery vector might we use?
To address DEC2 mutations, which affect neuronal circadian clock function and sleep-wake cycle regulations in the suprachiasmatic nucleus (and some other places), we would need to target brain tissue. And while AAVs and LNPs have their advantages when it comes to cargo capacity/lasting expression and affordability/immunogenicity, respectively, they both struggle to reach the brain with the efficiency needed to induce phenotypic changes in a living human. That is not to say this won’t ever be possible, but at least today, the vectors proven in humans have yet to meet the safety and efficacy thresholds to make them viable candidates for this hypothetical use case.
Given that lentivirus is best suited for ex vivo applications and that the brain is not one of those organs that can easily come out of the skull and be modified, it too is unsuitable. This leaves us with Herpes Simplex Virus.
The carrying capacity of HSV is sufficient to carry the full-length DEC2 gene, its tropism profile is ideal (meaning HSV naturally infects neurons and establishes latency in the nervous system) and its viral DNA tends to stick around, leading to persistent expression in the cells that govern sleep. Additionally, antigen testing for HSV antibodies would help avoid the toxic effects of an immune reaction. Of course, cost remains a concern, as producing the virus in pure batches at scale would likely entail tens to hundreds of thousands of dollars in manufacturing and labor alone. Still, based solely on technical merit, it could be a reasonable candidate for the delivery of DEC2, considering what has been shown to work in human patients today.
Given the many considerations at play in gene therapy, it’s clear why having numerous vector candidates on offer is so critical. A thriving gene therapy marketplace wouldn’t offer a single option but rather an array of offerings. As each of these platforms develops, so will new tools or hybrid approaches — combining the targeting precision of one system with the payload capacity of another, or the immunological advantages of non-viral systems with the efficiency of viral mechanisms.
As of this writing, the FDA lists 36 approved cell and gene therapies, with eight novel approvals having appeared in 2024 alone. In 2019, based on a pipeline of over 800 active investigational new drug applications, the FDA anticipated approving 10 to 20 new therapies annually by 2025. The impressive clinical successes achieved across existing methods suggest that the gene therapy field is only just getting started.
Eryney Marrogi is a medical student at the University of Vermont, with experience in biological engineering from working on mosquitoes at Harvard, AAV at Dyno Therapeutics, and novel biosensors at Caltech. Find him on Substack.
Thank you to Jacob Witten, Adrian Veres, Effie Klimi, Chris Stach, and Stephen Malina for providing feedback on this piece.
Illustrated by Ella Watkins-Dulaney.
Cite: Marrogi, E. “A Visual Guide to Gene Delivery.” Asimov Press (2025).
 | A guest post by
|
Thanks for this write-up!
I recently had the opportunity to review Eli Lilly's gene therapy pipeline while scoping a project for work. They had some billion-dollar acquisitions (Verve (LNP, Prevail (AAV)), and it looks like they are collecting the building blocks for gene therapy and successful delivery platforms; a big signal for this field. Worth adding to your table.
I work in clinical development and trials for gene therapy are fascinating!
One of the best breakdowns on this topic that I've come across. Would love one on the selection factors for cargo (e.g. gene therapy vs. editing vs. ASO, etc.)!